AiiDA by example: Computing a band structure
Contents
2. AiiDA by example: Computing a band structure#
Learning Objectives
In this section we will present a complete example of an AiiDA workflow, which defines the sequence of calculations needed to compute the band structure of silicon.
How to setup the input data and the details of the workflow execution will be discussed in subsequent sections. Here we simply give an initial overview of what it means to run an AiiDA workflow.
2.1. Interacting with AiiDA#
AiiDA can be controlled in two ways:
Using the
verdicommand line interface (CLI), or%verdimagic in Jupyter notebooks.Using the
aiidaPython API
For each project in AiiDA, we set up a profile, which defines the connection to the data storage, and other settings.
Show cell content
from local_module import load_temp_profile
data = load_temp_profile(
name="bands_workflow",
add_computer=True,
add_pw_code=True,
add_sssp=True,
add_structure_si=True,
)
data
Matplotlib is building the font cache; this may take a moment.
AiiDALoaded(profile=Profile<uuid='84a830b6c5d54d1cb9cdb2667d05969b' name='bands_workflow'>, computer=<Computer: local_direct (localhost), pk: 1>, code=<Code: Remote code 'pw.x' on local_direct, pk: 1, uuid: 43019f16-33cc-468a-99f3-b757d9b3b8bc>, pseudos=SsspFamily<1>, structure=<StructureData: uuid: 5071207a-7602-4223-a535-8a847351c70a (pk: 87)>, cpu_count=1, workdir=PosixPath('/home/docs/checkouts/readthedocs.org/user_builds/aiida-qe-demo/checkouts/latest/tutorial/local_module/_aiida_workdir/bands_workflow'), pwx_path=PosixPath('/home/docs/checkouts/readthedocs.org/user_builds/aiida-qe-demo/conda/latest/bin/pw.x'))
%verdi status --no-rmq
✔ version: AiiDA v2.0.4
✔ config: /home/docs/checkouts/readthedocs.org/user_builds/aiida-qe-demo/checkouts/latest/tutorial/local_module/_aiida_path/.aiida
✔ profile: bands_workflow
✔ storage: SqliteTemp storage [open], sandbox: /home/docs/checkouts/readthedocs.org/user_builds/aiida-qe-demo/checkouts/latest/tutorial/local_module/_aiida_path/.aiida/repository/bands_workflow
⏺ daemon: The daemon is not running
Within this profile, we have stored the initial input components for our workflow, including the pseudo-potentials, and the silicon structure:
%verdi storage info --detailed
Show cell output
entities:
Users:
count: 1
emails:
- user@email.com
Computers:
count: 1
labels:
- local_direct
Nodes:
count: 87
node_types:
- data.core.code.Code.
- data.core.structure.StructureData.
- data.pseudo.upf.UpfData.
process_types: []
Groups:
count: 1
type_strings:
- pseudo.family.sssp
Comments:
count: 0
Logs:
count: 0
Links:
count: 0
We have also set up the compute resource that we will use to run the calculations, and the code (pw.x) installed on that computer, which we will use to perform the electronic structure calculations.
Here, we will use our “local” machine to run the computations, but AiiDA can also be used to submit calculations to remote supercomputer schedulers, transporting data between the local machine and the remote computer.
%verdi computer show local_direct
Show cell output
--------------------------- -----------------------------------------------------------------------------------------------------------------------------------
Label local_direct
PK 1
UUID 9805a4c2-24d3-4120-9764-6cdea0c08202
Description local computer with direct scheduler
Hostname localhost
Transport type core.local
Scheduler type core.direct
Work directory /home/docs/checkouts/readthedocs.org/user_builds/aiida-qe-demo/checkouts/latest/tutorial/local_module/_aiida_workdir/bands_workflow
Shebang #!/bin/bash
Mpirun command mpirun -np {tot_num_mpiprocs}
Default #procs/machine 1
Default memory (kB)/machine
Prepend text
Append text
--------------------------- -----------------------------------------------------------------------------------------------------------------------------------
%verdi code show pw.x@local_direct
Show cell output
-------------------- ------------------------------------------------------------------------------------
PK 1
UUID 43019f16-33cc-468a-99f3-b757d9b3b8bc
Label pw.x
Description pw.x code on local computer
Default plugin quantumespresso.pw
Type remote
Remote machine local_direct
Remote absolute path /home/docs/checkouts/readthedocs.org/user_builds/aiida-qe-demo/conda/latest/bin/pw.x
Prepend text export OMP_NUM_THREADS=1
Append text
-------------------- ------------------------------------------------------------------------------------
2.2. Utilising a pre-defined workflow#
AiiDA plugins can declare workflow plugins, for use within AiiDA. These are workflows that are pre-defined, and can be used as-is, or as a starting point for your own workflows.
Here we utilise the quantumespresso.pw.bands workflow defined by the aiida-quantumespresso plugin.
%verdi plugin list aiida.workflows
Show cell output
Registered entry points for aiida.workflows:
* core.arithmetic.add_multiply
* core.arithmetic.multiply_add
* quantumespresso.matdyn.base
* quantumespresso.pdos
* quantumespresso.ph.base
* quantumespresso.pw.bands
* quantumespresso.pw.base
* quantumespresso.pw.relax
* quantumespresso.q2r.base
Report: Pass the entry point as an argument to display detailed information
%verdi plugin list aiida.workflows quantumespresso.pw.bands
Show cell output
Description:
Workchain to compute a band structure for a given structure using Quantum ESPRESSO pw.x.
The logic for the computation of various parameters for the BANDS step is as follows:
Number of bands:
One can specify the number of bands to be used in the BANDS step either directly through the input parameters
`bands.pw.parameters.SYSTEM.nbnd` or through `nbands_factor`. Note that specifying both is not allowed. When
neither is specified nothing will be set by the work chain and the default of Quantum ESPRESSO will end up being
used. If the `nbands_factor` is specified the maximum value of the following values will be used:
* `nbnd` of the preceding SCF calculation
* 0.5 * nspin * nelectrons * nbands_factor
* 0.5 * nspin * nelectrons + 4 * nspin
Kpoints:
There are three options; specify either an existing `KpointsData` through `bands_kpoints`, or specify the
`bands_kpoint_distance`, or specify neither. For the former those exact kpoints will be used for the BANDS step.
In the two other cases, the structure will first be normalized using SeekPath and the path along high-symmetry
k-points will be generated on that structure. The distance between kpoints for the path will be equal to that
of `bands_kpoints_distance` or the SeekPath default if not specified.
Inputs:
bands: required Data Inputs for the `PwBaseWorkChain` for the BANDS calculation.
scf: required Data Inputs for the `PwBaseWorkChain` for the SCF calculation.
structure: required StructureData The inputs structure.
bands_kpoints: optional KpointsData Explicit kpoints to use for the BANDS calculation. Specify either this or ` ...
bands_kpoints_distance: optional Float Minimum kpoints distance for the BANDS calculation. Specify either this or ...
clean_workdir: optional Bool If `True`, work directories of all called calculation will be cleaned at th ...
metadata: optional
nbands_factor: optional Float The number of bands for the BANDS calculation is that used for the SCF mult ...
relax: optional Data Inputs for the `PwRelaxWorkChain`, if not specified at all, the relaxation ...
Outputs:
band_parameters: required Dict The output parameters of the BANDS `PwBaseWorkChain`.
band_structure: required BandsData The computed band structure.
scf_parameters: required Dict The output parameters of the SCF `PwBaseWorkChain`.
primitive_structure: optional StructureData The normalized and primitivized structure for which the bands are computed.
seekpath_parameters: optional Dict The parameters used in the SeeKpath call to normalize the input or relaxed ...
Exit codes:
1: The process has failed with an unspecified error.
2: The process failed with legacy failure mode.
10: The process returned an invalid output.
11: The process did not register a required output.
201: Cannot specify both `nbands_factor` and `bands.pw.parameters.SYSTEM.nbnd`.
202: Cannot specify both `bands_kpoints` and `bands_kpoints_distance`.
401: The PwRelaxWorkChain sub process failed
402: The scf PwBasexWorkChain sub process failed
403: The bands PwBasexWorkChain sub process failed

The quantumespresso.pw.bands workflow provides a helpful method for setting up the default inputs for a given “protocol”, as to how fast/precise the calculation should be.
This provides a “builder” object, which stores all the inputs for the workflow.
from aiida_quantumespresso.workflows.pw.bands import PwBandsWorkChain
builder = PwBandsWorkChain.get_builder_from_protocol(
code=data.code,
structure=data.structure,
protocol="fast",
)
builder
Show cell output
Process class: PwBandsWorkChain
Inputs:
bands:
metadata: {}
pw:
code: pw.x code on local computer
metadata:
options:
max_wallclock_seconds: 43200
resources:
num_machines: 1
stash: {}
withmpi: true
parameters:
CONTROL:
calculation: scf
etot_conv_thr: 0.0002
forc_conv_thr: 0.001
tprnfor: true
tstress: true
ELECTRONS:
conv_thr: 8.0e-10
electron_maxstep: 80
mixing_beta: 0.4
SYSTEM:
degauss: 0.01
ecutrho: 240.0
ecutwfc: 30.0
nosym: false
occupations: smearing
smearing: cold
pseudos:
Si: ''
bands_kpoints_distance: 0.1
clean_workdir: true
metadata: {}
nbands_factor: 3.0
relax:
base:
kpoints_distance: 0.5
kpoints_force_parity: false
metadata: {}
pw:
code: pw.x code on local computer
metadata:
options:
max_wallclock_seconds: 43200
resources:
num_machines: 1
stash: {}
withmpi: true
parameters:
CELL:
cell_dofree: all
press_conv_thr: 0.5
CONTROL:
calculation: vc-relax
etot_conv_thr: 0.0002
forc_conv_thr: 0.001
tprnfor: true
tstress: true
ELECTRONS:
conv_thr: 8.0e-10
electron_maxstep: 80
mixing_beta: 0.4
SYSTEM:
degauss: 0.01
ecutrho: 240.0
ecutwfc: 30.0
nosym: false
occupations: smearing
smearing: cold
pseudos:
Si: ''
base_final_scf:
metadata: {}
pw:
metadata:
options:
stash: {}
pseudos: {}
max_meta_convergence_iterations: 5
meta_convergence: true
metadata: {}
volume_convergence: 0.05
scf:
kpoints_distance: 0.5
kpoints_force_parity: false
metadata: {}
pw:
code: pw.x code on local computer
metadata:
options:
max_wallclock_seconds: 43200
resources:
num_machines: 1
stash: {}
withmpi: true
parameters:
CONTROL:
calculation: scf
etot_conv_thr: 0.0002
forc_conv_thr: 0.001
tprnfor: true
tstress: true
ELECTRONS:
conv_thr: 8.0e-10
electron_maxstep: 80
mixing_beta: 0.4
SYSTEM:
degauss: 0.01
ecutrho: 240.0
ecutwfc: 30.0
nosym: false
occupations: smearing
smearing: cold
pseudos:
Si: ''
structure: Si
2.3. Running the workflow#
Workflows can be run in the interpreter using the run method, in a blocking manner, which we shall do here.
from aiida import engine
result = engine.run_get_node(builder)
result
Show cell output
Report: [104|PwBandsWorkChain|run_relax]: launching PwRelaxWorkChain<106>
Report: [106|PwRelaxWorkChain|run_relax]: launching PwBaseWorkChain<109>
Report: [109|PwBaseWorkChain|run_process]: launching PwCalculation<114> iteration #1
Report: [109|PwBaseWorkChain|results]: work chain completed after 1 iterations
Report: [109|PwBaseWorkChain|on_terminated]: remote folders will not be cleaned
Report: [106|PwRelaxWorkChain|inspect_relax]: after iteration 1 cell volume of relaxed structure is 40.97317396255211
Report: [106|PwRelaxWorkChain|run_relax]: launching PwBaseWorkChain<123>
Report: [123|PwBaseWorkChain|run_process]: launching PwCalculation<128> iteration #1
Report: [123|PwBaseWorkChain|results]: work chain completed after 1 iterations
Report: [123|PwBaseWorkChain|on_terminated]: remote folders will not be cleaned
Report: [106|PwRelaxWorkChain|inspect_relax]: after iteration 2 cell volume of relaxed structure is 41.15149425981942
Report: [106|PwRelaxWorkChain|inspect_relax]: relative cell volume difference 0.004352123109385916 smaller than threshold 0.05
Report: [106|PwRelaxWorkChain|results]: workchain completed after 2 iterations
Report: [106|PwRelaxWorkChain|on_terminated]: remote folders will not be cleaned
Report: [104|PwBandsWorkChain|run_scf]: launching PwBaseWorkChain<142> in scf mode
Report: [142|PwBaseWorkChain|run_process]: launching PwCalculation<147> iteration #1
Report: [142|PwBaseWorkChain|results]: work chain completed after 1 iterations
Report: [142|PwBaseWorkChain|on_terminated]: remote folders will not be cleaned
Report: [104|PwBandsWorkChain|run_bands]: launching PwBaseWorkChain<155> in bands mode
Report: [155|PwBaseWorkChain|run_process]: launching PwCalculation<158> iteration #1
Warning: c_bands: at least 1 eigenvalues not converged
Report: [155|PwBaseWorkChain|results]: work chain completed after 1 iterations
Report: [155|PwBaseWorkChain|on_terminated]: remote folders will not be cleaned
Report: [104|PwBandsWorkChain|results]: workchain succesfully completed
Report: [104|PwBandsWorkChain|on_terminated]: cleaned remote folders of calculations: 114 128 147 158
ResultAndNode(result={'primitive_structure': <StructureData: uuid: 0bd67e0d-319c-4d49-8efa-35735b92df46 (pk: 138)>, 'seekpath_parameters': <Dict: uuid: 0b6d72c7-e865-4930-9820-9473a962fb55 (pk: 136)>, 'scf_parameters': <Dict: uuid: 3797607c-478c-4f1a-9659-5fad2f031e41 (pk: 152)>, 'band_parameters': <Dict: uuid: 96cf1934-a4d7-4ace-b841-1e998a3d61e5 (pk: 163)>, 'band_structure': <BandsData: uuid: 43b15d67-4f3e-4b94-9d3a-cd15e5a8f558 (pk: 161)>}, node=<WorkChainNode: uuid: fa77f701-1902-4f28-a7a2-1fa001eec6ef (pk: 104) (aiida.workflows:quantumespresso.pw.bands)>)
Typically however, long running workflows are executed by using the submit method.
This will store the initial state of the workflow in the profile storage, and notify the AiiDA daemon to run the workflow in the background.
The AiiDA daemon can be launched using the verdi daemon start n command, with n being the number of worker processes to launch.
Each worker can asynchronously handle 1000s of individual calculations, allowing for a high-throughput of workflow submissions.

Each workflow and node stored in the AiiDA profile is assigned a unique identifier (a.k.a Primary Key), which can be used to reference them.
The execution of the workflows can be monitored using the verdi process list command, which will show the status of all running processes in the profile (or also finished ones with -a).
%verdi process list -a
Show cell output
PK Created Process label Process State Process status
---- --------- ---------------------------- --------------- ----------------
104 2m ago PwBandsWorkChain ⏹ Finished [0]
106 2m ago PwRelaxWorkChain ⏹ Finished [0]
109 2m ago PwBaseWorkChain ⏹ Finished [0]
110 2m ago create_kpoints_from_distance ⏹ Finished [0]
114 2m ago PwCalculation ⏹ Finished [0]
123 1m ago PwBaseWorkChain ⏹ Finished [0]
124 1m ago create_kpoints_from_distance ⏹ Finished [0]
128 1m ago PwCalculation ⏹ Finished [0]
135 29s ago seekpath_structure_analysis ⏹ Finished [0]
142 29s ago PwBaseWorkChain ⏹ Finished [0]
143 29s ago create_kpoints_from_distance ⏹ Finished [0]
147 28s ago PwCalculation ⏹ Finished [0]
155 21s ago PwBaseWorkChain ⏹ Finished [0]
158 21s ago PwCalculation ⏹ Finished [0]
Total results: 14
Report: last time an entry changed state: 0s ago (at 08:15:05 on 2022-10-04)
Warning: the daemon is not running
We can also monitor the progress of individual workflows using the verdi process status command, which will show the status of the individual steps of the workflow.
%verdi process status {result.node.pk}
PwBandsWorkChain<104> Finished [0] [7:results]
├── PwRelaxWorkChain<106> Finished [0] [3:results]
│ ├── PwBaseWorkChain<109> Finished [0] [4:results]
│ │ ├── create_kpoints_from_distance<110> Finished [0]
│ │ └── PwCalculation<114> Finished [0]
│ └── PwBaseWorkChain<123> Finished [0] [4:results]
│ ├── create_kpoints_from_distance<124> Finished [0]
│ └── PwCalculation<128> Finished [0]
├── seekpath_structure_analysis<135> Finished [0]
├── PwBaseWorkChain<142> Finished [0] [4:results]
│ ├── create_kpoints_from_distance<143> Finished [0]
│ └── PwCalculation<147> Finished [0]
└── PwBaseWorkChain<155> Finished [0] [4:results]
└── PwCalculation<158> Finished [0]
This work-chain demonstrates how we can build up a complex workflow from a series of individual calculations. In this case, the workflow is made up of the following steps:
The
PwRelaxWorkChainwill run multiple Quantum ESPRESSOvc-relaxcalculations, to make sure that there are no Pulay stresses present in the material and that the requested k-points density is respected in case there is a significant volume change in the material.Once the geometry has been optimized, SeeK-path will be used to primitivize and standardize the structure, as well as find the standard path along which to calculate the band structure.
A static calculation (
scf) is run to calculate the charge density for the structure obtained from SeeK-path.Finally, an NSCF is run to calculate the band structure along the path determined by Seek-path.
We shall also discuss in subsequent sections, how the PwBaseWorkChain can identify and recover from known failure modes, such as reaching the wall-time limit of the scheduler, or convergence failures.
2.4. Inspecting the results#
Once we the workflow has finished, we can inspect the results using the verdi process show command, which will show the results of the workflow, and its “attached” outputs.
%verdi process show {result.node.pk}
Show cell output
Property Value
----------- ------------------------------------
type PwBandsWorkChain
state Finished [0]
pk 104
uuid fa77f701-1902-4f28-a7a2-1fa001eec6ef
label
description
ctime 2022-10-04 08:12:54.952706+00:00
mtime 2022-10-04 08:15:05.401843+00:00
computer [1] local_direct
Inputs PK Type
----------------------------------- ---- -------------
bands
pw
pseudos
Si 52 UpfData
code 1 Code
parameters 99 Dict
max_iterations 100 Int
relax
base
pw
pseudos
Si 52 UpfData
code 1 Code
parameters 88 Dict
kpoints_distance 89 Float
kpoints_force_parity 90 Bool
max_iterations 91 Int
max_meta_convergence_iterations 92 Int
meta_convergence 93 Bool
volume_convergence 94 Float
scf
pw
pseudos
Si 52 UpfData
code 1 Code
parameters 95 Dict
kpoints_distance 96 Float
kpoints_force_parity 97 Bool
max_iterations 98 Int
bands_kpoints_distance 103 Float
clean_workdir 101 Bool
nbands_factor 102 Float
structure 87 StructureData
Outputs PK Type
------------------- ---- -------------
band_parameters 163 Dict
band_structure 161 BandsData
primitive_structure 138 StructureData
scf_parameters 152 Dict
seekpath_parameters 136 Dict
Called PK Type
-------- ---- ---------------------------
relax 106 PwRelaxWorkChain
seekpath 135 seekpath_structure_analysis
scf 142 PwBaseWorkChain
bands 155 PwBaseWorkChain
2.4.1. The provenance graph#
As well as storing the inputs and outputs of the workflow, and its composite calculations, AiiDA also stores the links between them, which can be used to reconstruct the provenance graph of the workflow.
This can be visualised using the verdi node graph generate command, or using the Graph Python API.
from aiida.tools.visualization import Graph
graph = Graph(graph_attr={"rankdir": "TB", "size": "8!,8!"})
graph.recurse_ancestors(result.node, annotate_links="both")
graph.recurse_descendants(result.node, annotate_links="both")
graph.graphviz

2.4.2. The output structure#
AiiDA’s StructureData class provides integration with both ASE, and Pymatgen, which can be used to inspect and visualise the structure.
pym_structure = result.node.outputs.primitive_structure.get_pymatgen()
pym_structure
Structure Summary
Lattice
abc : 3.8752542610912695 3.8752542610912695 3.8752542610912695
angles : 59.99999999999999 59.99999999999999 59.99999999999999
volume : 41.151494259818136
A : 0.0 2.7402185668397 2.7402185668397
B : 2.7402185668397 0.0 2.7402185668397
C : 2.7402185668397 2.7402185668397 0.0
PeriodicSite: Si (2.7402, 0.0000, 0.0000) [-0.5000, 0.5000, 0.5000]
PeriodicSite: Si (1.3701, 4.1103, 1.3701) [0.7500, -0.2500, 0.7500]
from ase.visualize.plot import plot_atoms
ase_atoms = result.node.outputs.primitive_structure.get_ase()
ax = plot_atoms(ase_atoms)
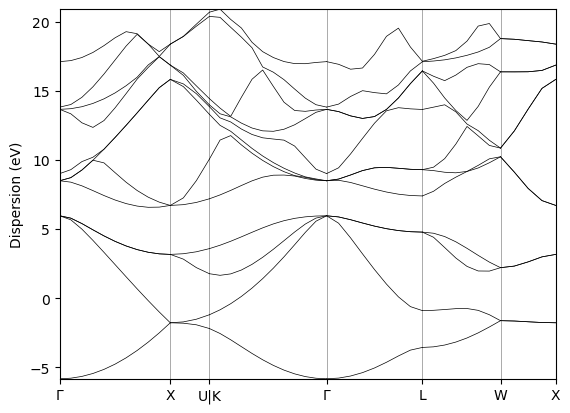
2.4.3. The output band structure#
Finally, we get to our desired result, the band structure of silicon computed using Quantum ESPRESSO 🎉
from local_module.bandstructure import plot_bandstructure
bands = result.node.outputs.band_structure
fig, ax = plot_bandstructure(bands)